
La crise du Covid-19 a mis en avant – entre autres – certains dysfonctionnements de notre système de santé et le dénuement dans lequel se trouve tous les jours le personnel des hôpitaux et des maisons de repos. Il est regrettable d’en être arrivé là pour apprécier à sa juste valeur leur travail ainsi que celui de toutes les « petites mains » tapies dans l’ombre que cette pandémie a enfin un peu mis en lumière.
Pour mieux comprendre certains des problèmes que nous avons connus pendant cette crise sanitaire, nous vous proposons ce trimestre de nous pencher sur l’univers de l’industrie pharmaceutique. De la conception d’un médicament à sa production à grande échelle, une série de procédés techniques et administratifs prenant une bonne décennie sont nécessaires. Nous allons détailler ces différentes étapes et essayer d’y voir plus clair dans cette entreprise qui génère des milliards d’euros et dont les valeurs éthiques ne semblent pas toujours à la hauteur de nos attentes. Bien que la littérature soit très complexe et très disséminée sur le sujet, nous avons essayé de vous en proposer une vision globale.
Concrètement, comment un médicament est réalisé et mis sur le marché 1 ?
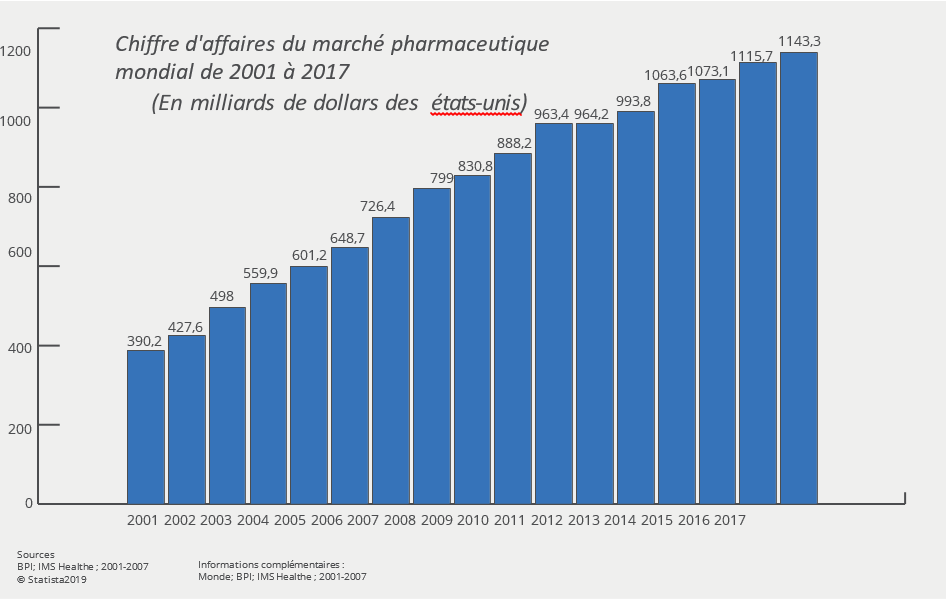
Un médicament est l’association d’un principe actif et d’excipients. Pour mettre au point un principe actif et vérifier son intérêt thérapeutique, il faut compter entre 10 à 15 ans de recherches, d’essais cliniques et de procédures administratives dans des domaines très divers comme la biologie, les biotechnologies, la chimie ou encore la galénique2. Toutes ces étapes nécessitent du personnel hautement qualifié et des investissements financiers importants. On parle d’environ 800 millions d’euros d’investissement, même plus d’un milliard selon certains groupes pharmaceutiques. Mais le jeu en vaut la chandelle comme l’indique le graphique ci-dessous.
Identifier une molécule3
C’est un travail de longue haleine mais extrêmement automatisé car il faut analyser entre 10.000 à 50.000 molécules pour avoir la chance d’en trouver une avec un effet thérapeutique réel. C’est aussi ce que l’on appelle la recherche exploratoire ou screening : les grands laboratoires pharmaceutiques
(Big Pharma4) mais également leurs sous-traitants vont soumettre des milliers de molécules à une batterie de tests systématiques pour étudier leurs propriétés chimiques et pharmacologiques. Ces molécules proviennent de molécules voisines de produits déjà utilisés comme principes actifs, d’extraction de plantes, de librairies5 de molécules ou encore via la modélisation de molécules par des logiciels spécifiques. Après ce screening, si une molécule semble avoir un intérêt thérapeutique, elle fait aussitôt l’objet d’un dépôt de brevet protégeant la molécule vis-à-vis de la concurrence.
Essais précliniques (environ 5 ans)
Pour vérifier l’intérêt thérapeutique des molécules, ces dernières sont testées in vitro sur des cellules animales. Ce travail permet de définir l’innocuité, la tolérance, l’efficacité… à des dosages différents. A ce niveau, nous passons de plusieurs dizaines de milliers à 100 molécules potentiellement intéressantes comme principes actifs.
Développement clinique (environ 5 ans)
Les chimistes et les spécialistes en biotechnologie sont déjà en action pour synthétiser les molécules qui ont passé les essais précliniques avec succès. A ce niveau, l’objectif est de synthétiser quelques grammes ou kilogrammes permettant de fournir la matière pour les essais suivants.
- Phase 1 = essais sur des volontaires sains et rémunérés, principalement pour vérifier les effets secondaires. Les médicaments à visée anticancéreuse ne sont pas testés en phase 1 et sont administrés directement à des patients malades.
- Phase 2 = patients malades. C’est une première validation qui permet de faire les essais à une plus grande échelle.
- Phase 3 = grands essais allant de 100 à plus de 1000 patients.
Ceci permet de mettre à jour d’éventuels effets secondaires non suspectés.
Rien n’est encore acquis à ce moment-là, car le moindre souci durant ces étapes implique l’arrêt immédiat des recherches. - Phase 4 ou pharmacovigilance = cette phase ne se déroule qu’une fois le médicament mis sur le marché. Il y a une surveillance permanente du produit tout au long de sa commercialisation.
Développement pharmaceutique
La conception d’un médicament n’est pas chose aisée. Les chercheurs doivent proposer – entre autres – des voies de synthèse suffisamment efficaces pour avoir des rendements intéressants, et surtout être capables de passer de la fabrication de quelques kilos en laboratoire à plusieurs tonnes en production6.
Il existe 3 façons de fabriquer un médicament :
- La synthèse totale par la chimie fine (issue de la chimie du pétrole) est la plus importante forme de création de molécules
- L’hémisynthèse qui correspond à la transformation chimique d’une molécule naturelle (obtenue par biotechnologie ou extraction)
- La biotechnologie (fermentation, extraction, réaction enzymatique)
A ce niveau, les sociétés pharmaceutiques elles-mêmes mais également un certain nombre de leurs sous-traitants sont sollicités pour synthétiser le principe actif. Les nouvelles molécules sont de plus en plus complexes et leur synthèse nécessite parfois plus d’une quinzaine d’étapes. Dans la pratique, il y a souvent plusieurs sous-traitants employés pour effectuer une ou plusieurs étapes. Un sous-traitant reçoit de son donneur d’ordre ou d’un autre sous-traitant une partie de molécule, il la modifie à son tour, et envoie ce nouvel intermédiaire au sous-traitant suivant, et ainsi de suite. Chacun réalise ainsi une ou plusieurs étapes pour arriver au produit final. De plus en plus fréquemment, le donneur d’ordre arrive en fin de parcours après avoir récupéré le principe actif fini ou quasi fini. Il réalise éventuellement la dernière synthèse et conditionne le principe actif avec les excipients choisis pour réaliser le médicament.
Pendant une bonne partie de ces procédés de synthèse, il est obligatoire de travailler en BPF (Bonnes Pratiques de Fabrication ou GMP en anglais). Ces BPF sont reprises dans un guide qualité (de plus de 400 pages) qui spécifie les bonnes pratiques à mettre en œuvre pour l’hygiène des locaux, le personnel, la gestion documentaire, les zones de stockage… Les documents réalisés à partir de ce guide, tout le long de la production, représentent des milliers de pages et seront audités par différents organismes et les sociétés donneuses d’ordre elles-mêmes. Ils serviront également pour le dépôt du dossier d’enregistrement.
Procédures administratives
Elles sont réalisées à différentes étapes d’avancement du processus.
>> Approbation des essais cliniques
Selon le règlement n°536/2014 du Parlement européen et du conseil du 16 avril 2014 relatif aux essais cliniques de médicaments à usage humain :
« Une demande d’autorisation d’essai clinique (DAEC) doit être déposée auprès des autorités nationales compétentes. Un Comité d’éthique de la recherche (REC) étudie également le protocole et donne un avis positif ou négatif. L’objectif est de s’assurer que la recherche respecte la dignité, les droits, la sécurité et le bienêtre des participants. Des essais cliniques sur des médicaments sont menés dans l’Union européenne (UE) conformément aux réglementations, directives et recommandations. La norme selon laquelle les essais cliniques sont menés est appelée « bonnes pratiques cliniques » ».
>> Les brevets
Les brevets sont des outils purement juridiques qui permettent de protéger les découvertes. Pour limiter la marge de manœuvre de la concurrence, les sociétés pharmaceutiques peuvent déposer plusieurs sortes de brevets :
- Le brevet sur la molécule. La plupart du temps le brevet ne concerne pas une molécule mais une famille de molécules
- Le brevet sur une ou plusieurs voies de synthèse ou brevet de procédé
- Le brevet pour les applications du médicament (ex : comme hypertenseur ou antidépresseur…)
- Le brevet galénique : c’est-à-dire la forme administrable (comprimé, injectable…)
- Le brevet sur la forme cristalline : un principe actif solide est en fait un agencement tridimensionnel de molécules. Un même produit peut avoir plusieurs agencements (formes cristallines) qui peuvent avoir des propriétés pharmacologiques différentes, comme sa vitesse de libérationdans l’organisme après ingestion…
- Le brevet sur des cocktails de molécules : dans le cas de la trithérapie par exemple
Tous ces brevets7 sont rédigés par des services juridiques spécialisés qui vont les soumettre aux différents pays susceptibles de fabriquer le médicament et donc de concurrencer le produit. En Europe, l’Office Européen des Brevets (OEB) protège les découvertes pour tout l’espace économique européen. Ces dépôts de brevets et la surveillance permanente de leur respect engendrent des coûts importants pour les entreprises pharmaceutiques. Cette protection est valable 20 ans avec une possibilité de prolongation jusqu’à 5 ans. Passé ce délai, le brevet tombe dans le domaine public et est donc libre d’exploitation.
Dès son dépôt, le brevet est accessible à tous. Il n’est pas scientifique dans le sens où vous ne trouverez pas la recette complète et explicite du produit. Cela permet malgré tout à la concurrence de voir ce qui est fait et de se préparer à proposer quelque chose de similaire.
En effet, certains « blockbusters 8 » représentent des chiffres d’affaires potentiels de plusieurs milliards de dollars. La concurrence est féroce. Vous avez compris qu’à part quelques initiés, nous sommes dans une jungle que nous ne soupçonnons pas.
>> Le dossier d’enregistrement9
Sans dossier d’enregistrement, tout ce qui a été fait avant ne sert à rien.
Seul ce dossier donne le sésame de l’autorisation de mise sur le marché (AMM) et donc de la commercialisation du médicament. Là encore, seule l’Europe a un organisme central, l’Agence Européenne des Médicaments (EMA), qui permet via une procédure centralisée de demander une AMM pour tous les pays de l’espace économique européen (31 Etats), à condition toutefois que la société ait une agence dans un des Etats membres. Si l’EMA refuse, il est encore possible de faire une demande par pays. C’est pourquoi il peut arriver, en Europe, que certains médicaments soient vendus dans certains pays et pas d’autres. Hors Europe, le dossier doit être déposé pays par pays. Les USA ont également un organisme central représentant tous leurs Etats : la Food and Drug Administration (FDA).
Evidemment, chaque demande d’AMM implique le paiement d’une redevance aux autorités sanitaires.
Le dossier d’enregistrement contient toute une série d’informations allant de l’historique de l’entreprise, des preuves des bonnes pratiques de fabrication, les procédés de synthèse, les impuretés, les analyses, les validations, la stabilité, la toxicologie… A la différence du brevet, seules certaines parties du dossier sont consultables par tous, les procédés de fabrication ne le sont pas.
>> La fixation des prix et le remboursement10
Pour qu’un médicament soit commercialisé à grande échelle et donc rentable pour la société qui l’a conçu, il doit être prescrit par les médecins et donc remboursé par la sécurité sociale. Pour cela, la société pharmaceutique doit soumettre un dossier de remboursement.
Chaque pays est souverain dans ce domaine. En Belgique, la demande est faite auprès de l’INAMI. Le
Ministre fédéral des affaires sociales examine les demandes d’admission au remboursement avec l’aide de la Commission de remboursement des médicaments (CRM).
Parallèlement, la société introduit une demande de prix auprès de la Commission des prix des spécialités pharmaceutiques (dépendant du Service Public Fédéral Economie).
Si tout convient, le médicament est inscrit sur la liste des spécialités pharmaceutiques remboursables et peut être commercialisé.
Le médicament générique
Une fois le brevet d’un médicament tombé dans le domaine public, son équivalent (générique) peut être produit par n’importe quelle société pharmaceutique. Toute la partie concernant les essais cliniques qui prend beaucoup de temps et d’argent n’est plus nécessaire, puisque déjà validée, seule la partie « synthèse de la molécule » est à créer ainsi que toute la partie documentaire correspondante.
En effet, les brevets initiaux ne dévoilent pas la voie de synthèse pour refaire la molécule et le dossier d’enregistrement, plus technique, n’est pas accessible à la concurrence. En résumé, si on compare à une recette de cuisine, vous avez la photo du plat, des bribes de la recette et quelques ingrédients. Les sociétés de génériques et leurs équipes de chercheurs en chimie et biotechnologies doivent donc mettre au point un nouveau procédé pour synthétiser la molécule (=principe actif). Pour être les premières sur le marché, elles sont déjà dans cette démarche avant que la molécule ne tombe dans le domaine public (5 à 7 ans avant). Le générique est fabriqué avec la même rigueur11 que le médicament original (même mieux car avec des procédés d’analyse plus récents donc plus performants). En résumé, le principe actif est le même que celui de marque, ainsi que le dosage et la forme galénique (un comprimé remplace un comprimé), seuls les excipients varient.
Le médicament générique commercialisé a un dossier d’enregistrement complet avec une étude de stabilité du produit, ainsi qu’une AMM et une demande de remboursement.
Le générique n’est pas un « sous médicament ». Il est réalisé dans les règles de l’art.
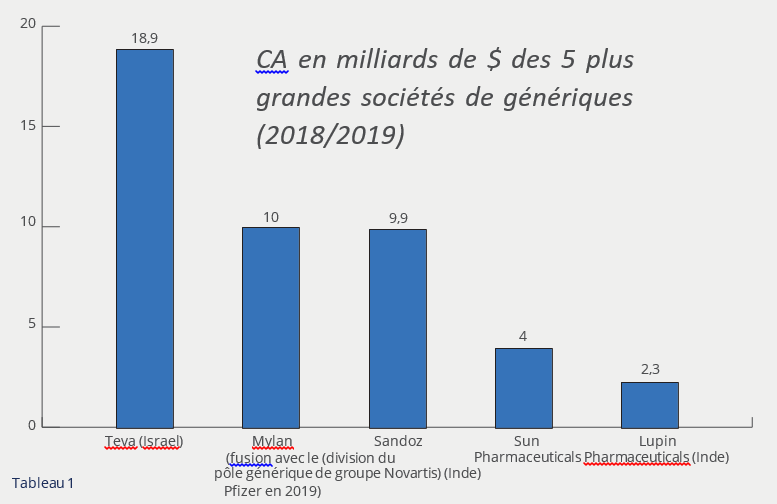
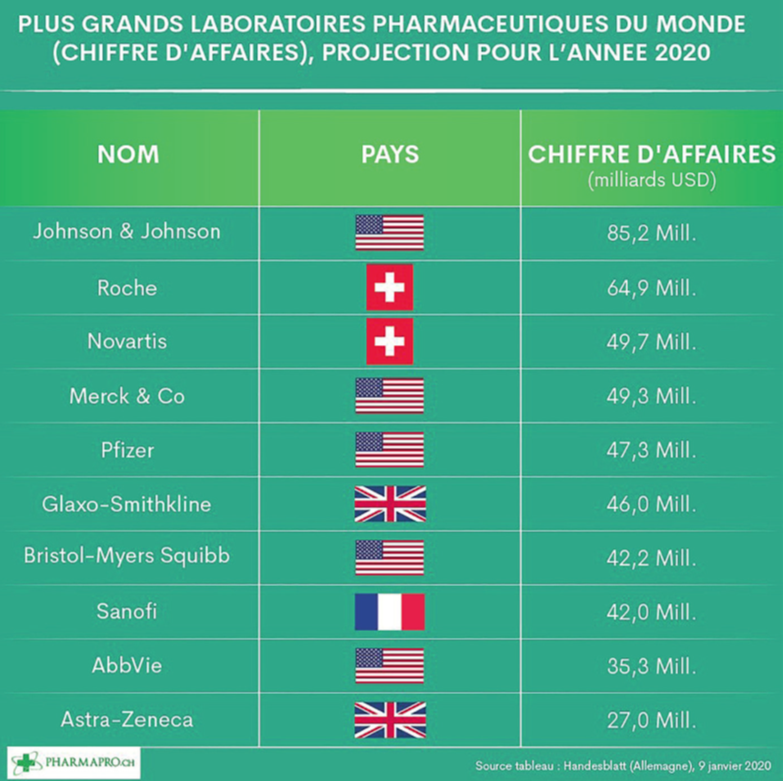
Bien que ces sociétés investissent moins (pas de brevets – sauf le brevet de procédés – et pas d’essais cliniques) que les sociétés de marque, elles gagnent aussi beaucoup moins d’argent. En effet, elles pratiquent des prix plus faibles exigés par les organismes d’assurance maladie, elles sont en concurrence avec les autres sociétés de génériques qui ont également pu sortir la même molécule et elles sont bien évidemment en concurrence12 avec des nouvelles molécules (mais pas forcément innovantes appelées molécules de 2ème génération) mises sur le marché par les Big Pharma. Leur chiffre d’affaires13 (tableau 1) est sans commune mesure avec ces dernières14 (tableau 2).
Les Contract Research Organization (CRO)/ Contract ManufacturingOrganization (CMO)15
Comme nous l’avons signalé précédemment, les Big Pharma font appel tout le long de la recherche et développement (essais cliniques, découverte de voies de synthèse performantes…) à des sous-traitants appelés CRO (ou SRC société de recherche contractuelle en français) et plus tard à des sous-traitants CMO, c’est-à- dire des façonniers qui fabriquent les produits.
Il est donc impossible pour le consommateur de savoir où est réellement conçu et surtout fabriqué un médicament. Il y a beaucoup d’intermédiaires dans la chaîne de production car la synthèse est fragmentée. En général, le principe actif arrive dans l’entreprise pharmaceutique donneuse d’ordre
(Big Pharma ou générique, en Europe ou aux USA) qui réalise la formulation16 avec les excipients adéquats et le packaging. Le produit est stocké et distribué directement aux pharmacies ou aux grossistes. Cette multiplication d’intervenants permet entre autres aux Big Pharma de faire de substantiels bénéfices (les CRO et CMO européens ou hors UE sont mis sous la pression et les exigences des Big Pharma qui font des marges colossales), mais également de dispatcher les responsabilités en cas de problèmes.
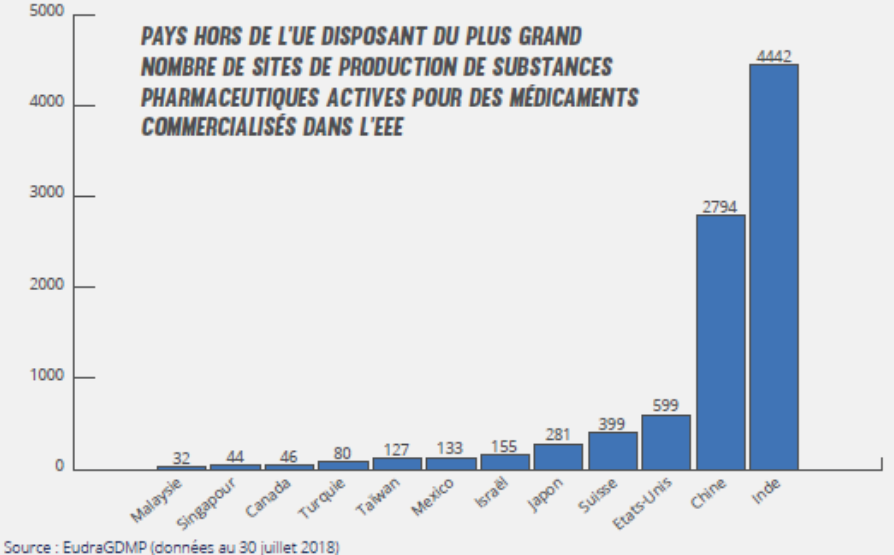
Sites de production17
Il existe des milliers de sites de production à travers le monde et la plus grande partie se trouve en Asie. Selon un rapport du Sénat18 français de 2018 : « Le syndicat de l’industrie chimique organique de synthèse et de la biochimie (Sicos) précise toutefois que les sites français produisant des substances pharmaceutiques actives peuvent ne pas mettre en œuvre toutes les étapes de la chaîne de fabrication mais seulement les dernières d’entre elles et donc s’approvisionner en intermédiaires et dans tous les cas en matières premières de base. Ces matières ne sont aujourd’hui plus toutes disponibles en Europe mais bien souvent uniquement en Asie. Sans une filière chimie forte dans notre pays, ou au moins en Europe, il est peu probable in fine que toutes les MPUP (Matières premières à usage pharmaceutique) puissent être fabriquées en France ».
40% de produits finis (le médicament) et 80% des principes actifs utilisés en Europe sont élaborés hors de l’UE. Avant la mondialisation, il y a 30 ans, 80% des principes actifs étaient produits en Europe.
Nous sommes face à un défi important car l’Europe entière a un problème de dépendance sanitaire. Elle ne peut absolument pas garantir aux patients de ses Etats la capacité à réagir en cas de problème.
La Belgique investit beaucoup dans le domaine pharmaceutique. Elle s’engage dans les laboratoires de recherches publics mais également dans le privé en aidant financièrement les industries pharmaceutiques à coup de subsides, d’exonérations de salaires ou de taxes19. La stratégie belge est clairement en faveur d’un maillage d’entreprises et de Start-up fort sur son territoire. En 2016, ce secteur représentait plus de 16 milliards de CA, plus de 26.000 équivalents temps plein et 137 entreprises. Malgré tous les avantages que la Belgique offre, elle est logée à la même enseigne que tous les pays d’Europe. De plus, il n’y a pas d’uniformisation des prix des médicaments pour l’Europe. En cas de coup dur, c’est celui qui fera le plus gros chèque qui sera servi le premier. La Belgique et ses 11 millions d’habitants ne seront probablement pas prioritaires.
Par contre, ses infrastructures et le personnel hautement qualifié dont la Belgique bénéficie peuvent lui permettre de prendre sa part sur un marché européen qui doit évoluer vers plus d’indépendance.
Les pénuries20, 21, 22
Depuis plus d’une dizaine d’années déjà, la littérature spécialisée alerte les médias et les services publics concernés des aléas d’approvisionnement en médicaments aussi bien pour les officines que pour les pharmacies d’établissement de santé. En Belgique, en France, dans toute l’Europe, et même en Amérique du Nord, la pénurie ou l’indisponibilité de certains médicaments est de plus en plus criante. Rien que dans le Royaume de Belgique près de 500 produits ont été signalés manquants en 2019.
Ces pénuries impliquent évidemment que les patients ne reçoivent plus les traitements adéquats. Selon Alain Astier, professeur en pharmacie témoignant pour Arte23, « en milieu hospitalier, le traitement peut être suspendu ou remplacé par une alternative moins efficace ou moins bien adaptée à la pathologie. Certaines publications ont démontré une réelle augmentation de la mortalité chez des patients qui ont été traités via une alternative, notamment dans des cas de chimiothérapies ».
Les anesthésiants viennent aussi à manquer régulièrement. En plus des risques encourus par les patients, les pharmaciens hospitaliers ou d’officines passent leur temps à jongler avec les fournisseurs (même si le système des appels d’offre ne le facilite pas) pour trouver des substituts, quand ils existent. Il faut compter entre 6 mois à plus d’un an et demi pour réaliser une campagne de production d’un médicament (cela peut aller jusqu’à plusieurs années pour certains vaccins). Il est donc difficile de fournir rapidement un médicament quand la production a été arrêtée.
Un phénomène multifactoriel :
>> La chaîne de production et sa cascade de sociétés intermédiaires est un véritable souci. En effet, un grain de sable dans les rouages et c’est toute la chaîne qui s’arrête de fonctionner. Ces grains de sable sont nombreux :
- En cas de problème lié à la qualité du produit fourni (impuretés trop importantes, non-respect des BPF, présence d’impuretés génotoxiques…). Lors d’audits, le conseil de l’Europe suspend ou retire des certificats de conformité (CEP), dont les 3/4 concernent la Chine et l’Inde. Ces suspensions ou retraits rompent l’approvisionnement en produits intermédiaires ou en principes actifs. Mais ces contrôles qualité ne sont pas suffisants. Ils sont cinq fois plus nombreux en
Europe qu’en Chine ou en Inde, alors que ces deux pays sont les fournisseurs mondiaux. A eux deux, ils possèdent près d’un millier de sites de fabrication de principes actifs et plusieurs milliers de sites d’intermédiaires de synthèse. Sur tous ces sites, seulement une centaine ont été inspectés. Il est totalement illusoire de croire que des contrôles sont réalisés en suffisance. Les sociétés européennes ou américaines qui récupèrent les produits de ces pays doivent absolument faire de sérieuses analyses. Si les contrôles s’avèrent mauvais, c’est toute la production qui s’arrête.
Le scandale du moment est la découverte des N-nitrosamines (mutagènes cancérigènes), qui sont des impuretés trouvées dans toute la famille des sartans (un des antihypertenseurs les plus vendus au monde).
- En plus du principe actif ou des intermédiaires de synthèse, les excipients peuvent également avoir des soucis de qualité.
- Les risques naturels. Dans les années 2000, un tsunami au Japon a mis à l’arrêt une cinquantaine de sites pharmaceutiques.
- Des ruptures de stocks liées à une organisation de travail à flux tendu. Pour leur rentabilité, les sociétés ne peuvent pas se permettre de stocker des produits.
>> L’augmentation de la demande mondiale en médicaments
>> L’arrêt de la production de produits devenus non rentables. Si la société pharmaceutique décide de poursuivre la production, elle est d’office totalement produite en Chine ou en Inde. C’est pourquoi beaucoup d’antibiotiques ou d’autres molécules simples comme le paracétamol ou l’ibuprofène ne sont plus du tout fabriqués en Europe.
>> Les risques géopolitiques, si des problèmes surviennent avec la Chine ou l’Inde, nous ne sommes pas en position de force pour négocier.
Quelques solutions24, 25
Depuis quelques années, l’Europe et différents groupes de travail se penchent sur ces pénuries. Malheureusement l’industrie pharmaceutique représente plus de 1000 milliards de chiffres d’affaires (en 2019)26 et leur lobby est tellement puissant que même les Etats27 ont du mal à se faire entendre.
Pourtant quelques solutions sont proposées :
- Recréer les conditions d’une production pharmaceutique locale (une chaîne plus complète qu’aujourd’hui). Attention, certaines sociétés affirment pouvoir produire des centaines de médicaments différents. En réalité, ces sociétés ont soit des produits finis qu’elles mettent en boîte, soit le principe actif qu’elles formulent pour arriver au produit final. En général, elles ne fabriquent pas le principe actif, et pour être réactives vis-à-vis de la demande, elles doivent avoir beaucoup de stocks mais à un moment ou un autre elles dépendront des intermédiaires fabriqués hors UE.
- Réaliser un programme public de production et distribution pour les médicaments essentiels qui sont devenus non rentables et dont les productions sont arrêtées par les entreprises pharmaceutiques.
- Etre plus exigeant vis-à-vis de l’industrie pharmaceutique qui ne doit pas avoir pour seule ambition son chiffre d’affaires.
- Perler, c’est-à-dire vendre de manière déconditionnée, pour limiter le gaspillage de médicaments.
- Faire des achats groupés entre Etats membres. Même si cette possibilité existe, elle ne semble pas encore être activée.
En résumé, l’univers de l’industrie pharmaceutique est d’une très grande complexité. Depuis une trentaine d’années, début de la mondialisation, nous avons assisté à un éclatement de la conception et de la production de médicaments à travers le monde. Cette organisation a permis aux entreprises pharmaceutiques de faire des profits conséquents. Mais ce système arrive à ses limites. En 30 ans, en Europe, nous sommes passés de 80% de fabrication de principes actifs à 20%, au profit de l’Asie, nous mettant face à une dépendance sanitaire inquiétante. Bien que l’industrie pharmaceutique soit soumise à une réglementation stricte, il devient de moins en moins évident de garantir la qualité des produits et conséquemment leur production. Vu le nombre d’intermédiaires et leur localisation, le contrôle efficace des sites est quasi impossible. Si l’Europe peut se targuer d’avoir des chercheurs et des entreprises performantes dans le domaine, elle est totalement dépendante des matières premières venues hors UE quand ce n’est pas tout simplement des produits finaux. Il est temps qu’elle reprenne les rênes de son indépendance avec les conséquences que cela engendrera.
Béatrice Touaux
Bibliographie
- « Le secteur pharmaceutique en Belgique » par Christophe Goethals, Marcus Wunderle, « Courrier Hebdomadaire du CRISP » 2018
- La forme galénique d’un médicament concerne son aspect (comprimé, injectable…) et son type d’absorption (libération lente, gastrorésistant…)
- https://leem.org/recherche-et-developpement
- multinationales de l’industrie pharmaceutique
- Librairies de molécules ou banques de molécules
- Appelé aussi scale up ou la montée en échelle. De la synthèse en laboratoire à la production industrielle il y a un monde de difficultés.
- « Le secteur pharmaceutique en Belgique » par Christophe Goethals, Marcus Wunderle, « Courrier Hebdomadaire du CRISP » 2018
- Signifie « qui fait exploser le quartier », plus couramment utilisé pour les films, en réalité ce terme concerne tous les produits appelés à avoir un grand succès (le « viagra » est un blockbuster)
- « Procédure centralisée », Académie européenne des patients – EUPATI
- « Le secteur pharmaceutique en Belgique » par Christophe Goethals, Marcus Wunderle, « Courrier Hebdomadaire du CRISP » 2018
- « Les génériques fonctionnent aussi bien que les originaux (étude) », Pharmapro.ch
- « Les stratégies des pharmas pour retarder la commercialisation de génériques » par Natalie Bougeard, RTS info
- « Top five generic drug makers » par Victoria Rees, European Pharmaceutical Review
- « Plus grands laboratoires pharmaceutiques du monde (chiffre d’affaires) » par Xavier Gruffat, Pharmapro.ch
- « Le secteur pharmaceutique en Belgique » par Christophe Goethals, Marcus Wunderle, « Courrier Hebdomadaire du CRISP » 2018
- Formulation = mélange du principe actif et des excipients pour créer le médicament sous une forme galénique (comprimé, gélule…)
- « L’industrie pharmaceutique : ses projets de développement, leurs caractéristiques et leur management » par Didier Gourc, Sophie Bougaret
- « Pénuries de médicaments et de vaccins : renforcer l’éthique de santé publique dans la chaîne du médicament », rapport d’information n°737 sept. 2018, Sénat (France)
- « Le secteur pharmaceutique en Belgique » par Christophe Goethals, Marcus Wunderle, « Courrier Hebdomadaire du CRISP » 2018
- « Pénurie de médicaments : il faut relocaliser la production en Europe », Tribune dans le Journal du Dimanche, 2019
- « Pénurie de médicaments en Belgique : il suffit ! », Carte blanche dans Le Soir, 2019
- « Médicaments : Ruptures de stocks, ruptures d’approvisionnement », Académie Nationale de Pharmacie (France)
- « Pénurie de médicaments : l’Europe en quête de solutions », Arte TV, 2019
- « Pénuries de médicaments et de vaccins : renforcer l’éthique de santé publique dans la chaîne du médicament », rapport d’information n°737 sept. 2018, Sénat (France)
- « Matières premières pharmaceutiques, mondialisation et santé publique », Académie Nationale de Pharmacie (France)
- « 1000 milliards d’euros de profits en vingt ans : comment les labos sont devenus des monstres financiers », par Olivier Petitjean, bastamag.net, 2019
- « Lobbying : comment l’industrie pharmaceutique prend d’« assaut » les institutions européennes», par Simon Gouin, bastamag.net, 2019


